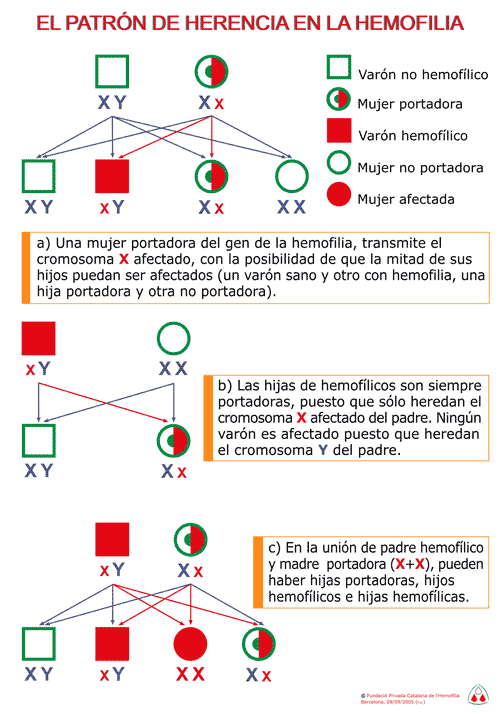
En esta figura se ilustra la descendencia familiar de
un enfermo hemofílico y la de una mujer portadora.
- En el primer caso todos los hijos varones son normales
y todas las hijas son portadores.
- En el segundo caso la mitad de las hijas son portadoras
y la mitad de los hijos presentan la enfermedad
1.2. Tipos de hemofilia:Se han descrito los déficits
congénitos de diferentes factores de la coagulación
(factor II, factor V, factor VII, factor X, fibrinógeno),
pero identificamos con el nombre de hemofilia A la deficiencia
de factor VIII, y con el de hemofilia B (llamada también
enfermedad de Christmas) la de factor IX. La hemofilia
A es entre 4 y 5 veces más frecuente que la B.
1.3. Gravedad de la hemofilia:Se han descrito tres
niveles de gravedad de la hemofilia (grave, moderada
y leve) en relación a la actividad de factor
presente en el plasma. Este hecho es muy importante
por dos motivos:
Generalmente, pero no siempre, el
nivel de actividad de factor presente en el plasma del
enfermo determina la frecuencia y gravedad de las manifestaciones
hemorrágicas, y como consecuencia define las
necesidades del tratamiento.
De este modo, cuanto menor sea el
nivel de actividad de factor en el plasma, mayores serán
las hemorragias y también aumentará el
uso de factor para tratarlas.
El nivel de actividad de factor,
y por tanto la gravedad de la hemofilia, se mantiene
constante en una familia determinada. De esta manera
el niño hemofílico en una familia con
historia de hemofilia moderada también tendrá
una forma moderada de esta enfermedad.
Cuando el nivel de factor es inferior
al 1%, hablamos de hemofilia grave. En estos casos las
hemorragias espontáneas son muy frecuentes y
graves.
Frecuentemente los enfermos con hemofilia
grave presentan alteraciones importantes en las articulaciones
como consecuencia de las repetidas hemorragias articulares.
Las pequeñas cantidades de
factor, entre el 1 y el 5%, presentes en los enfermos
con hemofilia moderada son suficientes para proporcionar
una cierta protección, de manera que las hemorragias
espontáneas son menos graves y menos frecuentes.
Los enfermos con hemofilia leve tienen
niveles de factor entre el 5 y el 25%. Estos no presentan
hemorragias espontáneas y frecuentemente se les
diagnostica cuando son adultos.
1.4. La coagulación
normal:
La hemostasia es el conjunto
de mecanismos y reacciones que tienen como finalidad
mantener la sangre en el interior de los vasos sanguíneos,
impidiendo su salida a los tejidos que la envuelven.
Este fenómeno tiene 4 fases: fase vascular, fase
plaquetaria, fase plasmática y fibrinólisis.
Fase vascular:
Consiste en un reflejo nervioso
que tiene como objetivo la liberación de unas
sustancias que provocan la contracción del vaso
lesionado, disminuyendo el flujo de sangre y en consecuencia
la hemorragia. Fase plaquetaria
Se desarrolla en dos etapas:
la adhesión de las plaquetas al subendotelio
vascular y la agregación de las plaquetas entre
sí. En esta fase es muy importante la participación
del factor von Willebrand, que establece puntos de unión
entre las plaquetas y los vasos lesionados.
El factor von Willebrand es una proteína
del plasma a la que está fijado el factor VIII.
Su deficiencia o ausencia da lugar a otra enfermedad
hemorrágica conocida como enfermedad de von Willebrand.
Otras proteínas como la fibronectina
y el fibrinógeno así como el calcio son
también necesarios para el buen funcionamiento
de este mecanismo.
Fase plasmática:
Es el conjunto de reacciones enzimáticas
que tienen la finalidad de convertir el fibrinógeno,
que es una proteína soluble, en fibrina insoluble.
La fibrina tiene una estructura fibrilar y es la base
del coágulo.
El conjunto de enzimas que intervienen
en esta fase son los llamados factores de coagulación,
que circulan en la sangre de forma inactiva y se convierten
en formas activas por la acción de otro factor
previamente activado.
En la figura se observan las diversas
recciones que tienen lugar en esta fase, que se desencadenan
por dos vías:
- Intrínseca, cuando la sangre entra en contacto
con el colágeno subendotelial y un
conjunto de proteínas que forman
lo que se denomina sistema de contacto.
La cefalina es la prueba de laboratorio
que analiza esta vía.
- Extrínseca, que se inicia cuando la sangre
entra en contacto con el factor tisular, que es una
proteína presente en los tejidos y que se libera
cuando hay una lesión vascular. La prueba de
coagulación que explora la vía extrínseca
es el tiempo de Quick o tasa de protrombina.
(falta img)
Fibrinólisis:
Al mismo tiempo que se desencadenan
las reacciones descritas, se ponen en funcionamiento los
mecanismos que tienden a disolver el coágulo con
la finalidad de que el vaso lesionado vuelva a ser permeable
y la sangre circule nuevamente. Este mecanismo se denomina
fibrinólisis y se basa fundamentalmente en la actividad
de una enzima, la plasmina, que degrada la fibrina.
Del perfecto equilibrio entre estos mecanismos
depende que la sangre se mantenga líquida en
el interior de los vasos sanguíneos, que únicamente
se forme un coágulo cuando se produce una lesión
vascular y que posteriormente los vasos vuelvan a ser
permeables.
2. Manifestaciones
clínicas de la hemofilia
Las manifestaciones hemorrágicas
del enfermo hemofílico están en relación
con la gravedad de la hemofilia, presentándose
de forma espontánea en los hemofílicos
severos y sólo después de traumatismos
o intervenciones quirúrgicas en los enfermos
moderados.
Les hemorragias pueden aparecer a cualquier
edad o en cualquier parte del organismo. Su gravedad
dependerá del volumen de la hemorragia, de su
localización y de la capacidad de dejar secuelas
o lesiones irreversibles.
2.1. Las hemorragias
en el bebé
Hasta un 2% de los enfermos pueden
presentar hemorragia intracraneal durante el parto.
En alguna ocasión, esta puede ser la forma de
presentación de la enfermedad, especialmente
cuando no existen antecedentes.
Cuando se hace un diagnóstico prenatal
se debe evitar la vacuoextracción (extracción
mediante ventosa) del feto, dado que los hematomas cefálicos
pueden producir anemia y tardan en reabsorberse.
A veces pueden asociarse a hemorragia cerebral.
Durante los primeros meses, las manifestaciones clínicas
son poco importantes.
Si es necesario practicar análisis,
las extracciones tienen que hacerse con el máximo
cuidado para evitar la aparición de hematomas
importantes.
Los enfermos deben recibir todas las vacunas
propias de la edad y, además, las de la hepatitis
A y B. Con la dentición pueden aparecer sangrados
bucales, pero la complicación oral más
frecuente es el sangrado por la rotura del frenillo
de la lengua durante la fase de desarrollo oral.
Al empezar a caminar, los hematomas de
las piernas y de los glúteos son los más
frecuentes y empiezan a aparecer las hemartrosis.
2.2. Localizaciones hemorrágicas
más frecuentes
Hemorragias mucosas:
Pese a que generalmente revisten poca gravedad,
generalmente requieren tratamiento. Las más frecuentes
son las epistaxis (hemorragia nasal), las gingivorragias
(hemorragia por las encías), las secundarias
a heridas por mordedura de la lengua o a la caída
de la primera dentición (dientes de leche).
Especial gravedad pueden tener las secundarias a heridas
por espinas de pescado en la faringe ya que pueden producir
hematoma faríngeo con obstrucción de las
vías aéreas, y en consecuencia con la
imposibilidad de respirar normalmente. Hematomas
musculares:
Se producen espontáneamente, por
traumatismos directos o por esguinces en ejercicios
violentos. Se ponen de manifiesto por dolor, aumento
del volumen y del calor local y impotencia funcional.
Las lesiones musculares provocadas por
la presencia de sangre crean una tendencia a producir
nuevas hemorragias del músculo dañado.
Dado que hay músculos encapsulados,
la tensión provocada por la sangre vertida puede
producir la destrucción del músculo, la
compresión del sistema vascular (arterias y venas)
y de los nervios vecinos con consecuencias funcionales
importantes.
Esto se conoce con el nombre de síndrome
compartimental, que se produce fundamentalmente en el
antebrazo y en la pierna.
Cabe destacar el hematoma del psoa que se manifiesta
por dolor en la ingle y flexión del muslo, a
menudo tiene tendencia a reaparecer.
Hemorragia cerebral:
Puede producirse de manera espontánea,
si bien generalmente aparece después de traumatismos.
Se manifiesta por la presencia de dolor de cabeza, vómitos
y pérdida del nivel de conciencia.
En ocasiones, con deficiencias motoras y trastornos de
la coordinación. Requieren tratamiento substitutivo
inmediato con el factor deficiente y en ocasiones puede
ser necesario también el tratamiento quirúrgico.
Hemorragia articular:
Son las más frecuentes en los enfermos
hemofílicos. Se caracterizan por dolor, aumento
del tamaño de la articulación, calor y pérdida
de movimiento.
La articulación afectada queda inmovilizada en
la posición que permita un mayor volúmen
de la cavidad articular. La presencia de sangre
en el interior de la articulación pone en marcha
una serie de mecanismos que afectan a la membrana sinovial,
provocando su hipertrofia y la tendencia a presentar
nuevas hemorragias.
Los depósitos de hierro y la inflamación
crónica de la sinovial (sinovitis crónica
hipertrófica) afectará también
al cartílago y al hueso. A la larga se producirá
una destrucción de la articulación con
pérdida de movilidad e incluso su fijación
en posiciones viciosas.
Para evitar estas complicaciones, es muy importante
el tratamiento correcto de las hemorragias articulares.
Hemorragia urinaria:
Las hematurias espontáneas no son
infrecuentes en el enfermo hemofílico. Generalmente,
no es necesario un tratamiento específico, el
reposo y una abundante hidratación son suficientes.
En ocasiones, cuando aparecen de forma secundaria a
la presencia de litiasis (cálculo o piedra),
es necesario un tratamiento urológico específico.
Hemorragia digestiva:
Son producidas por lesiones digestivas diversas
típicamente hemorrágicas:
- hernia de hiato,
- ulcus,
- varices esofágicas,
- tumores y enfermedades ulcerativas como la enfermedad
de Crohn y la colitis ulcerosa.
Se ponen de manifiesto por la presencia de hematemesis
(vómitos de sangre), melenas (sangre oscura en
la deposición) y rectorragias (sangre roja en
la deposición).
Además del tratamiento substitutivo, con factor,
y si es necesario también con sangre, es indispensable
diagnosticar el foco hemorrágico, generalmente
mediante endoscopia.
Hemoptisis:
La aparición de hemoptisis no es más
frecuente en los enfermos hemofílicos pero pueden
ser más graves. Son secundarias a diferentes patologías
del aparato respiratorio (tuberculosis, tumores, fístulas
artiovenosas y otras alteraciones bronquiales.
En estos casos será necesario diagnosticar
la lesión responsable de la hemorragia mediante
broncoscopia o exámenes radiográficos.
En todos los casos es necesario iniciar
precozmente el tratamiento con el factor deficiente
para normalizar la coagulación. Se debe acudir
al hospital de forma urgente en las situaciones siguientes:
- Hemorragias que por su localización puedan
comportar importantes pérdidas sanguíneas
que requieran la transfusión de sangre.
En este apartado se incluyen las hemorragias
digestivas, las hemorragias en la cavidad abdominal
y los hematomas en grandes músculos.
- Hemorragias localizadas en zonas vitales: cavidad
craneal y vías aéreas.
2.3. Diagnóstico
de la hemofilia:
Se puede pensar o sospechar en el diagnóstico
de hemofilia ante enfermos con hemorragias espontáneas
o secundarias a traumatismos, especialmente si aparecen
en las primeras etapas de la vida o en el momento del
nacimiento.
La evaluación detallada de los antecedentes
hemorrágicos del enfermo y de su familia será
una ayuda importante para orientar el correcto diagnóstico
de hemofilia.
A continuación es indispensable
la participación del laboratorio. En los enfermos
con coagulopatías congénitas se evidencian
alteraciones en las pruebas que miden globalmente la
coagulación.
Concretamente en el caso de la hemofilia
A y B se encuentra alargada la cefalina. La confirmación
del tipo de hemofilia se obtiene cuando se detecta una
ausencia o una disminución significativa del
factor VIII (hemofilia A) o del factor IX (hemofilia
B).
2.4. Diagnóstico
molecular de la hemofilia:
La hemofilia se manifiesta cuando en los
genes de los factores VIII o IX se producen mutaciones,
es decir, alteraciones en el DNA que dan lugar a la
ausencia o pérdida de funcionalidad de la proteína.
Ya que los genes constituyen el material
que se transmite de padres a hijos, estas mutaciones,
y por lo tanto la enfermedad, también son transmitidas
a la descendencia.
El diagnóstico molecular de una
enfermedad genética como la hemofilia consiste
en la identificación del defecto o mutación
en el gen o bien en la identificación del cromosoma
que es portador del gen defectuoso.
Esto tiene una repercusión directa
ya que posibilita la realización de un adecuado
consejo genético y diagnóstico prenatal
a los miembros de la familia que así lo soliciten.
La identificación del cromosoma
portador del gen mutado se realiza mediante los estudios
de ligamiento: se utilizan marcadores polimórficos
cercanos a los genes responsables que permiten relacionar
un cromosoma determinado de la familia estudiada con
la manifestación de la enfermedad.
Estas técnicas, sin embargo, tienen
una serie de limitaciones que condicionan su uso, como
pueden ser los casos esporádicos, las familias
no informativas para los polimorfismos o la ausencia
de al menos un familiar hemofílico vivo.
Gracias a los recientes avances en la tecnología
de secuenciación del DNA, y para evitar las limitaciones
intrínsecas del análisis de ligamiento,
se han diseñado protocolos simplificados para
poder detectar la presencia de mutaciones en los genes
de los factores VIII y IX.
En el caso de la hemofilia A existe una
mutación que es mucho más frecuente que
el resto, es la llamada inversión del intrón
22. Esta inversión es responsable del 50% de
las hemofilias A severas.
Por lo tanto, un primer paso en el diagnóstico
molecular de la hemofilia A severa es ver si esta inversión
es la responsable de la enfermedad en la familia.
Las técnicas empleadas para detectar
esta mutación no requieren la secuenciación
y por lo tanto pueden ser realizadas de manera más
o menos rápida.
El resto de mutaciones descritas causantes
de hemofilia A y B incluyen mutaciones puntuales, delecciones,
inserciones y están localizadas a lo largo de
los genes.
Esto obliga a determinar toda la secuencia
o composición del gen del factor de coagulación
correspondiente en el enfermo y compararla con una secuencia
normal para identificar las mutaciones presentes.
Las mutaciones encontradas, como ya se
ha mencionado, permiten el diagnóstico prenatal
y consejo genético adecuados, pero además,
estas mutaciones son enviadas a bases de datos internacionales
conjuntamente con datos clínicos, de manera que
constituyen una fuente de estudio que permitirá
en el futuro introducir mejoras en las terapias de sustitución
con factores recombinantes así como en los ensayos
de terapia génica.
3. Tratamiento
de la hemofilia
3.1. Concentrados de factores de coagulación
Los aspectos que permiten diferenciar los
diferentes concentrados de factores de coagulación
son: la fuente biológica de obtención
(productos derivados del plasma y productos recombinantes),
la pureza y los métodos de inactivación
viral incluidos en el proceso de producción.
Los concentrados plasmáticos se
obtienen después de fraccionar, purificar y someter
el plasma a técnicas específicas de inactivación
viral.
La calidad y seguridad del producto final
son consecuencia de la suma de diversas etapas que incluyen
la selección de donantes, el análisis
de las donaciones individuales y del plasma para fraccionamiento
y las etapas específicas de inactivación
viral.
Los concentrados de FVIII derivados del
plasma se clasifican según su grado de pureza
en productos de pureza intermedia (PI), alta pureza
(AP) y ultrapuros (UP).
La mayoría de estos productos contienen
una cantidad relevante de albúmina, que se utiliza
como estabilizante.
Algunos de los preparados contienen factor
von Willebrand funcional, por este motivo son utilizados
también en el tratamiento de la enfermedad de
Von Willebrand.
Los productos de tecnología recombinante
se obtienen después de diversas etapas:
1. introducción del gen que codifica el factor
VIII o IX en células de mamíferos,
2. multiplicación de estas para obtener un banco
de células con capacidad
de formar factor VIII o IX,
3. incubación de las células en un medio
de cultivo apropiado para que produzcan
el factor deseado,
4. purificación del factor producido por estas
células,
5. tratamiento para la inactivación viral,
6. estabilización del factor y liofilización.
Los productos recombinantes tienen
un elevado grado de pureza.
Dependiendo del proceso de producción
utilizado, pueden contener pequeñas cantidades
de proteína y ADN de la célula huésped,
trazas de inmunoglobulinas murinas como consecuencia
de la purificación del factor con anticuerpos
monoclonales murinos y de albúmina humana o bovina
utilizadas en el medio de cultivo.
Actualmente se tiende a la utilización
de medios de cultivo sin proteínas de origen
humano o animal y a la substitución de la albúmina
humana por sacarosa como estabilizante del producto
final.
En
las tablas I, II y III se recogen los diferentes factores
de coagulación
actualmente disponibles en nuestro país. Tabla
I. Concentrados de factor VIII
PRODUCTO Laboratorio PROCEDENCIA PURIFICA-CIÓN
INACTIVA-CIÓN VIRAL ESTABILI-ZACIÓN INDICACIÓN
HAEMATE P®Aventis Plasma humano Precipitación
Pasteuriza-ción Albúminahumana Hemofilia
A Enfermedad devon Willebrand.
BERIATE P®Aventis Plasma humano Cromatografíaint.
Iónico Pasteuriza-ción Sacarosa Hemofilia
A.
FANHDI®Grifols Plasma humano Cromatografíaafinidad
heparina S/D +80 ºC, 72h Albúminahumana
Hemofilia A.
HEMOFIL®Baxter Plasma humano CromatografíaInmunoafinidad
S/D Albúminahumana Hemofilia A.
MONOCLATE P®Aventis Plasma humano CromatografíaInmunoafinidad
Pasteuriza-ción Albúminahumana Hemofilia
A.
RECOMBINATE®Baxter Recombinante(CHO) Cromatografías
int. iónico yinmunoafinidad - Albúminahumana
Hemofilia A.
REFACTO®Wyeth Recombinante(CHO) Cromatografíasint.
iónico yinmunoafinidad S/D Sacarosa Hemofilia
A.
KOGENATE BAYER® BayerHELIXATE NEXGEN® Aventis
Recombinante(BHK) Cromatografías Int. Iónico
yInmunoafinidad S/D Sacarosa Hemofilia A
BHK= células renales cría hámster;
CHO= células ovario hámster xino;
S/D=solvente/detergente
1 Actividad específica excluyendo albúmina
y FvW
2Molécula de FVIII con delección del dominio
B
Tabla II. Concentrados
de factor IX y complejo de protrombina
PRODUCTO Laboratorio, PROCEDENCIA, PURIFICACIÓN,
INACTIVACIÓNVIRAL, ESTABILIZACIÓN, INDICACIÓN
FACTOR IX-X PBEHRINGÒ
Aventis Plasma humano Adsorciónsobre resina Pasteurización
Estabilizantesno proteicos Déficit FIX, FX.
IMMUNINESTIM PLUSÒ
(FIX)Baxter Plasma humano Cromatografíaint. iónico
yint. Hidrofóbica Vapora presión +Detergente
Estabilitzantesno proteicos Hemofilia B.
MONONINEÒ
(FIX)Aventis Plasma humano Cromatografía Inmunoafinidad
Tiocianato sódico+ Ultrafiltración Estabilitzantesno
proteicos Hemofilia B.
BENEFIXÒ
(FIX)Baxter Recombinante(CHO) Cromatografíaint.
Iónico Nanofiltración Estabilitzantesno
proteicos Hemofilia B
Tabla III. Otros concentrados de factores de coagulación
PRODUCTO Laboratorio
PROCE-DENCIA PURIFICA-CIÓN INACTIVA-CIÓN
VIRAL ESTABILIZA-CIÓN INDICACIÓN
FEIBA IMMUNO TIM
4Ò(complejo protrombina activado)Baxter PlasmaHumano
AdsorciónSobre resina Vapora presión Estabilizantesno
proteicos Hemofilia A y Bcon inhibidor.
NOVOSEVENÒNovo
Nordisk Recombinante(BHK) Cromatato-grafíaint.
Iónico yinmunoafi-nidad Detergente Estabilitzantesno
proteicos Hemofilia A y Bcon inhibidor
3.2. Administración del factor
En primer lugar, es necesario seguir escrupulosamente
las instrucciones del fabricante del factor que están
descritas en el prospecto del producto. Como normas
generales se tiene que tener en cuenta:
- Reservar el producto liofilizado (el frasco con polvo
blanco) en la nevera a 4º C, no en
el congelador. Los otros elementos (agua
destilada, aguja y jeringa) no es necesario
que estén en estas condiciones.
- Preparar el factor en una superficie limpia y que
después, si se precisa, se pueda
limpiar con lejía.
- Desinfectar la superficie de la piel con una solución
desinfectante.
- Tirar las agujas y jeringas en un contenedor rígido.
3.3. Modalidades de tratamiento
y dosis
El tratamiento substitutivo se puede hacer
de forma profiláctica o sólo cuando se
produce hemorragia.
En este último caso, es necesario
aplicarlo lo más rápidamente posible.
Si se administra antes de las primeras 4 horas, la respuesta
es más efectiva y rápida.
Por esta razón se aconseja el tratamiento
domiciliario. La dosis y duración del tratamiento
dependerá de la gravedad de la hemorragia.
En la tabla se exponen las guías
generales para tratar las hemorragias más frecuentes.
Estos datos deben ser considerados como aproximados,
ya que en cada caso es necesario valorar aspectos individuales
de cada enfermo.
HEMORRAGIA NIVEL ÓPTIMO
FACTOR % DURACIÓN TRATAMIENTO
días:
Articular |
30 - 50
% |
1 - 3 días |
Hematoma muscular |
30 - 50 " |
1 - 3 " |
Digestiva |
30 - 50 " |
2 - 3 " |
Faringe |
40 - 60 " |
3 - 4 " |
Retroperitoneal |
30 - 50 " |
3 - 4 " |
Intracranial |
60 - 80 " |
7 - 10 " |
Urinaria |
30 - 50 " |
1 - 2 " |
Menor |
20 - 30 " |
1 - 2 " |
Los concentrados de factor se pueden
administrar en forma de bol (dosis única repetida
según las necesidades) o en infusión continua.
Esta última modalidad se utiliza
cuando se tienen que mantener niveles plasmáticos
altos de factor durante un período alargado,
tal y como pasa en las hemorragias agudas graves o durante
las intervenciones quirúrgicas.
El tratamiento profiláctico se aplica
a enfermos con deficiencia grave (factor VIII o IX inferior
al 1%) para evitar la aparición de artropatía
secundaria a las hemartrosis repetidas.
Se empieza en los primeros años de
vida y consiste en la administración de tres
dosis semanales en hemofilia A y dos dosis en hemofilia
B para mantener niveles mínimos superiores al
1-2% de forma permanente. Las dosis oscilan entre 30
y 50 UI por kg de peso.
Muchas veces, empezar este tratamiento tan
pronto es difícil porque no se dispone de accesos
venosos adecuados y es necesario implantarlos, solución
no exenta de complicaciones, especialmente las infecciones.
3.4. Otros tratamientos
A veces, un análogo de la vasopresina,
el acetato de desmopresina (DDAVP o Minurinâ),
administrado por vía nasal o intravenosa, puede
ser útil para la profilaxis o para el tratamiento
de las hemorragias leves en enfermos con hemofilia A
moderada o leve.
Este fármaco provoca un aumento inmediato
pero transitorio de los niveles de factor VIII y no
tiene efectos secundarios notables, exceptuando rubor
facial y conjuntival transitorios.
Los antifibrinolíticos (ácido
tranexámico o Amchafibrinâ y ácido
épsilo-amino-caproico o Caproaminâ) son
una buena opción como tratamiento coadyuvante
a las hemorragias de mucosas, exceptuando las hematurias.
Se administran por vía oral o intravenosa y son
muy bien tolerados.
Existen también hemostáticos
por vía tópica como la trombina liofilizada,
colágeno y otros, que no han demostrado demasiada
eficacia.
En cambio, otras substancias preparadas
comercialmente (Tisucol®) son capaces de producir
un coágulo de fibrina y son útiles al
aplicarlas como hemostáticos sobre las heridas.
3.5. Complicaciones del tratamiento con factores
de coagulación
Actualmente el nivel de seguridad en relación
a la transmisión de virus patógenos conocidos
es muy elevada como consecuencia de todas las medidas
que se han establecido en los últimos años,
y que se han descrito en el apartado anterior.
En este sentido es necesario destacar que
el objetivo principal del desarrollo de nuevos productos
es la minimización del riesgo de transmisión
de enfermedades.
Aunque en los últimos años
se ha producido una cierta alarma en relación
a la posible transmisión de la variante de la
enfermedad de Creutzfeldt-Jakob por los factores, en
base a los conocimientos actuales se puede afirmar que
este riesgo es muy bajo o inexistente.
La formación de inhibidores, continúa
siendo hoy en día una de las complicaciones más
graves del tratamiento de la hemofilia.
Aunque en un principio se creyó que
la utilización de productos recombinantes se
asociaba a una mayor incidencia de inhibidores, los
estudios recientes indican que su aparición es
parecida en los dos tipos de factor, plasmático
y recombinante.
Otras complicaciones del tratamiento con
factores de coagulación, como pueden ser las
alteraciones de la función inmune o las complicaciones
tromboembólicas, han sido muy minimizadas con
la utilización de productos de elevada pureza.
3.6. Los inhibidores
Los inhibidores del factor VIII y IX son
anticuerpos que aparecen en algunos enfermos hemofílicos
después de recibir concentrados del factor correspondiente.
Estos anticuerpos son capaces de neutralizar
el factor administrado condicionando que el tratamiento
sea ineficaz.
En la actualidad es la complicación
más grave de los enfermos hemofílicos,
ya que dificulta mucho la prevención y el tratamiento
de las hemorragias.
Aparecen en aproximadamente el 25% de los
enfermos con hemofilia A y en el 5% de los afectados
de hemofilia B.
Generalmente, se producen después
de las primeras administraciones de factor, apareciendo
en enfermos muy jóvenes.
Actualmente se sabe que el tipo de mutación
del paciente constituye un factor de riesgo para la
formación de inhibidores, y se ha detectado una
mayor incidencia de inhibidores asociada a determinadas
mutaciones del gen del FVIII.
Cabe sospechar la presencia de un inhibidor
cuando la respuesta clínica al tratamiento no
es la esperada.
Aunque la eficacia del tratamiento sea correcta,
es recomendable hacer la determinación periódica
de la presencia de inhibidores mediante análisis
de laboratorio, basados en la capacidad que tiene el
plasma del enfermo de neutralizar, en el laboratorio,
el factor VIII o el factor IX.
La actividad de los inhibidores se mide
en Unidades Bethesda (UB).
Una UB/ml es la actividad de un inhibidor
capaz de neutralizar 0.5 unidades de factor VIII o factor
IX.
Se consideran inhibidores de baja respuesta
a los que no superan las 10 UB, y de alta respuesta
a los que tienen una actividad superior a las 10 UB.
3.7. Alternativas terapéuticas en los enfermos
hemofílicos con inhibidor. Anulación del
inhibidor:
Cuando el título del inhibidor es
bajo, se pueden utilizar dosis más altas de factor
VIII o factor IX para el tratamiento de las hemorragias.
El factor VIII porcino tiene las mismas
calidades hemostáticas que el factor VIII humano
y en algunos enfermos no es reconocido por el inhibidor.
Puede producir efectos secundarios, como
reacciones alérgicas y trombocitopenia, aunque
han mejorado con los nuevos métodos de purificación;
actualmente se utiliza poco.
Los concentrados de complejo de protrombina
activada han demostrado su eficacia terapéutica,
aunque tampoco están exentos de efectos secundarios
al utilizarlos en dosis elevadas (por ejemplo, trombosis).
Se utilizan en dosis de 50-100 UI por kg cada 8-12 horas.
Desde hace unos años, el factor VIIa
recombinante es otra alternativa terapéutica
muy eficaz. Su inconveniente es su corta vida media
en el plasma, lo que requiere administraciones cada
2-3 horas para el control de las hemorragias.
Tal y como pasa en los enfermos sin inhibidor,
es necesario utilizar medidas complementarias como reposo,
inmovilización, aplicación de frío
local, etc.
Dado que las alternativas terapéuticas
en los enfermos con inhibidor son más limitadas,
una vez diagnosticada la presencia del inhibidor, es
necesario plantearse su anulación; es el llamado
tratamiento de inmunotolerancia.
Consiste en administrar inyecciones repetidas
(generalmente diarias) del factor de coagulación
que falta (VIII o IX) con la finalidad de que el sistema
inmunitario del propio enfermo se vuelva tolerante al
factor deficitario, efecto que se consigue en un 75-85%
de los enfermos tratados.
4. Otras
recomendaciones
4.1. Higiene oral
Es evidente que en los enfermos con coagulopatías
las intervenciones odontológicas, incluidas las
más senzillas o rutinarias como las obturaciones
o empastes dentales, representan un problema a tener
muy en cuenta dado el riesgo hemorrágico.
Por este motivo, los enfermos hemofílicos,
a menudo, se encuentran con dificultades para ser atendidos
en las consultas convencionales.
Como consecuencia de estos hechos, la prevención
de las principales patologías orales, caries
y enfermedad periodontal (gingivitis y periodontitis)
en el enfermo hemofílico adquiere una transcendencia
muy superior a la de las demás personas.
Podemos asegurar que si en la población
normal, sin patologías previas, esta prevención
es importante, en el enfermo hemofílico, especialmente
si coexisten infecciones víricas, pasa a ser
un objetivo fundamental.
Las patologías que hemos mencionado
anteriormente son altamente susceptibles a las medidas
preventivas. Las principales medidas preventivas de
la cavidad oral están basadas en los hábitos
de higiene oral diarios.
Es importante destacar que no se ha demostrado
hasta el momento que la hemofilia predisponga a sufrir
con más severidad caries o gingivitis (inflamación
de las encías).
Normalmente, en el enfermo hemofílico
así como en los otros enfermos odontológicos,
el sangrado de las encías se produce más
frecuentemente por la inflamación de los tejidos
gingivales, consecuencia de la falta de un cepillado
eficaz, que por la alteración de la coagulación
coexistente.
Como norma general, los niños y
los adultos han de hacer un mínimo de dos cepillados
dentales diarios, por la mañana y por la noche,
de tres minutos de duración en cada caso, utilizando
una pasta dental con flúor.
El uso de soluciones específicas
a base de flúor u otras sustancias que refuercen
la salud de las encías serán indicadas
únicamente por el profesional a cada enfermo
en particular según la edad y la patología
presente.
Es necesario aconsejar también una
visita periódica al odontólogo. Semestral
o anual, dependiendo de la edad del enfermo y del riesgo
detectado de alguna de las principales enfermedades
orales.
En aquellos casos en que sea necesario
hacer un tratamiento en la cavidad oral, es indispensable
que exista una buena comunicación entre el hematólogo
y el odontólogo para establecer un protocolo
de actuación que tenga en cuenta el riesgo de
la intervención y la posible necesidad de administrar
factor.
Desde un punto de vista odontológico,
se consideran de riesgo las intervenciones siguientes:
- la limpieza dental que requiere profundizar por debajo
de las encías,
- la anestesia troncular, utilizada con frecuencia en
el tratamiento de los molares inferiores,
- las extracciones dentales (exodoncia).
Las obturaciones o empastes dentales son
intervenciones de bajo riesgo, siempre que no requieran
un grado de anestesia profunda.
Aunque la colocación de prótesis
dentales y aparatos de ortodoncia no son manipulaciones
de riesgo, es necesario hacer el control adecuado de
los aparatos con la finalidad de prevenir la aparición
de úlceras y lesiones que conlleven hematomas
u otras hemorragias en la cavidad oral.
4.2. Ejercicio físico
La práctica de ejercicio físico
y deporte es importante en el enfermo con hemofilia
como una forma amena de mantener el tono muscular que
protege las articulaciones. Además es también
una excelente forma de relación social con otros
niños o adultos.
En primer lugar es fundamental escoger
una actividad satisfactoria para cada enfermo y que
su práctica no suponga un riesgo superior al
posible beneficio.
Como norma general, no debemos someter las
articulaciones con lesiones previas, a movilizaciones
que fuerzen el arco articular o a resistencias que provoquen
una sobrecarga.
La potenciación muscular, básica
para la prevención de la patología articular,
se puede obtener, sin complicaciones, mediante la práctica
de ejercicios isométricos, que contraen el músculo
sin movilizar la articulación adyacente.
Naturalmente es posible ir más allá
y en este sentido la Federación Mundial de Hemofilia
aconseja los siguientes deportes:
- Natación
- Tenis mesa (ping-pong)
- Senderismo
- Pesca
- Baile
- Golf
- Bolos
- Badminton
- Ciclismo
- Paddle
Por norma general, se consideran peligrosos
todos los deportes que conllevan una cierta violencia
(boxeo, motociclismo) o contacto físico (balonmano,
baloncesto).
Aunque el fútbol es el deporte que
más se practica, especialmente en las escuelas,
no es recomendable dado el riesgo de contacto directo
con la pelota y el continuo contacto, a menudo violento,
entre los jugadores.
Antes de iniciar una actividad deportiva,
es necesario hacer un período de calentamiento
para prevenir lesiones musculares, y al finalizar se
completa la sesión con suaves ejercicios de estiramiento.
Es importante utilizar un calzado adecuado
que controle bien el pie y el impacto del talón.
En caso de molestias articulares o musculares,
por pequeñas que sean, es necesario interrumpir
inmediatamente la actividad. Si existen lesiones articulares
o musculares, es indispensable esperar a su completa
resolución antes de iniciar nuevamente la actividad
deportiva.
Los enfermos que están en tratamiento
profiláctico pueden aprovechar los períodos
con buenos niveles de factor para desarrollar estas
actividades deportivas, con menos riesgo de presentar
complicaciones hemorrágicas.
En la práctica de un deporte
por parte de los enfermos con hemofilia, es necesario
encontrar un equilibrio entre el beneficio esperado,
como consecuencia de la actividad física y la
potenciación muscular, y las posibles complicaciones.
Será necesario seleccionar una actividad
bastante atractiva para el enfermo pero que se adapte
a la posible patología articular existente.
Dado que la mayoría de los enfermos
en edad de crecimiento siguen pautas de tratamiento
profiláctico, es recomendable coordinar el tratamiento
y la práctica de deporte, de manera que la máxima
actividad coincida con los niveles más altos
de factor.
5. La
terapia génica en hemofilia
La terapia génica es una manera nueva
de tratar las enfermedades basada en la introducción
de material genético dentro de las células
para corregir un problema biológico.
Los genes son fragmentos de ADN que contienen
la información para la formación de las
proteínas necesarias para el funcionamiento normal
de todo el organismo.
La terapia génica pretende corregir
la enfermedad proporcionando a las células información
para que sean ellas mismas las que fabriquen el producto
activo (la proteína), en lugar de administrarlo
directamente como se hace con los fármacos convencionales.
En el caso de la hemofilia se administraría
al enfermo el gen del factor de coagulación que
le falta en lugar de darle el factor.
Aunque en el futuro será posible
reparar los errores que existen en un gen y dan lugar
a la enfermedad, los objetivos actuales son más
modestos.
Actualmente, la terapia génica trata
de corregir el defecto en la función de un gen
anómalo introduciendo una copia normal de este
en las células.
La investigación actual está
centrada en desarrollar genes más eficaces, y
vehículos o vectores que se encarguen de hacer
que el material genético entre dentro de las
células.
Los vectores más eficaces para que la entrada
de material genético sea en cantidad suficiente
son los de tipo viral.
En ellos se eliminan los genes fundamentales
para su multiplicación y se sustituyen por el
gen terapéutico para aprovechar su capacidad
de entrar dentro de las células.
Hasta el momento se han realitzado cuatro
ensayos clínicos en humanos, en los que se han
utilizado diferentes estrategias. De los resultados
obtenidos en ellos se ha podido deducir que las estrategias
utilizadas y las dosis suministradas son seguras.
En el primer ensayo, que tuvo lugar hace
10 años en la China, se trataron a dos hermanos
con hemofilia B grave con células propias previamente
extraídas y a las que se había introducido
el gen del factor IX.
Así se consiguió una persistencia
de la actividad de más de un año. Hasta
noviembre de 1998 no se realizó el siguiente
ensayo, en esta ocasión fue con la hemofilia
A.
La empresa Transkaryotic Therapy, Inc trató
a los enfermos con células propias en las que
se había introducido el gen del factor VIII,
esta vez sin la ayuda de vectores virales.
Los resultados que se han publicado este
año muestran que con esta estrategia se pueden
conseguir niveles de entre el 1-2% respecto a los normales.
A mediados de 1999 se iniciaron dos ensayos
más, en estos casos aplicando los vectores virales
directamente en los pacientes.
La empresa Chiron trabajó en la administración
intravenosa de un tipo de virus con el gen del factor
FVIII, mientras que la empresa Avigen en su ensayo utilizó
la inyección intramuscular de otro virus, más
pequeño y con el gen del factor FIX. En los dos
casos se ha observado una disminución de la frecuencia
de sangrado.
En el año 2000 se han iniciado dos
ensayos más de los que no se dispone aún
de resultados.
6. 10
puntos clave en la hemofilia
01. Hay actividades que no te convienen, disfruta de
todo aquello que puedes hacer.
02. Aprende a administrarte el factor siguiendo las
medidas higiénicas necesarias, esto te
dará autonomía.
03. El tratamiento precoz de las hemorragias puede evitar
complicaciones más serias; no te quedes nunca
sin factor en casa.
04. Ante la presencia de hemorragias que puedan ser
vitales (cerebrales y faringe)ponte factor y acude ;rápidamente
al Hospital; consulta con el médico ante cualquier
situación de duda.
05. No tomes aspirina ni otros antiagregantes plaquetares,
consulta con tu médico qué fármacos
puedes tomar.
06. Practica de manera habitual el ejercicio físico
que te pueda ser más útil.
07. Sé muy cuidadoso con la higiene oral y acude
al dentista una vez al año.
08. Es muy importante realizar estudios genéticos
para conocer la existencia de otras portadoras en la
familia.
09. El consejo genético es muy aconsejable ante
el planteamiento de cualquier nuevo embarazo en la familia.
10. En caso de desplazamiento averigua cuáles
son los posibles hospitales de referencia.
7. Hospitales
de referencia y centros de tratamiento de Cataluña
Unidad de Hemofilia de
la Vall d’Hebron
Ciudad Sanitaria de la Vall d’Hebron
Hospital de Traumatología y Rehabilitación
Paseo de la Vall d’Hebron, s/n
08035 Barcelona
tf. 93 274 62 07
Servicio de Hematología
Hospital de la Santa Creu i Sant Pau
Av. Sant Antoni Mª Claret, 167
08025 Barcelona
tf. 93 347 31 33
Servicio de Hemoterapia
y Hemostasia
Hospital Clínic i Provincial de Barcelona
Casanovas, 143
08036 Barcelona
tf. 93 227 54 00
Hospital de Sant Joan
de Déu (BCN)
Ctra. de Esplugas, s/n
08034 Barcelona
tf. 93 280 40 00
Residencia Sanitaria
Josep Trueta
Avda. de França, s/n
17005 Girona
tf. 972 20 27 00
Residencia Sanitaria
Joan XXIII
Mallofré i Guasch, s/n
43007 Tarragona
tf. 977 21 77 50
Residencia Sanitaria
Arnau de Vilanova
Huesca, s/n
25001 Lleida
tf. 973 24 81 00
Resposable del Àrea d'informació mèdica:
Dr. Miquel Rutllant Bañeres, nomenat pel Patronat de la FPCH, número de col·legiat 4679, especialitat hematologia i hemoteràpia. |